转录水平解析昆虫病原线虫共生菌菌株新型杀菌蛋白PPIA-L20基因表达调控——材料与方法
1 材料与方法
1.1 试验材料
伯氏致病杆菌Xenorhabdus bovienii NN6菌株是本实验室从斯氏线虫Steinernema oregonense侵染致死的大腊螟血腔中分离鉴定的,菌种保存于实验室-80 ℃超低温冰箱中。尖孢镰刀菌西瓜专化型(Fusariurn oxysporum f. sp. niveum)的1号生理小种,由江苏省农业科学院蔬菜所瓜类组提供。NBTA鉴别培养基:酵母提取物3 g·L-1,胰蛋白胨5 g·L-1,营养琼脂(NA)20 g·L-1,2,3,5-三苯基氯化四氮唑(TTC)0.1 g·L-1,溴百里酚蓝(BTB)0.025 g·L-1,丙酮酸钠(SP)1 g·L-1。LB+SP液体培养基:酵母提取物5 g·L-1,胰蛋白胨10 g·L-1,NaCl 10 g·L-1,丙酮酸钠(SP)1 g·L-1。YSG液体培养基:甘油6.9 g·L-1,大豆蛋白胨25.17 g·L-1,MgSO4·7H2O 1.57 g·L-1,(NH4)2SO4 2.55 g·L-1,KH2PO4 0.87 g·L-1,K2HPO4 1.11 g·L-1,Na2SO4 1.81 g·L-1。马铃薯培养基(PDA):马铃薯200 g·L-1,葡萄糖20 g·L-1,琼脂15~18 g·L-1。PSA培养基:马铃薯蔗糖培养基粉末46 g·L-1。
1.2 试验方法
1.2.1 共生菌的活化与发酵生长曲线的测定
从-80 ℃超低温冰箱取出菌种在NBTA平板上划线培养,72 h后接种蓝绿色单菌落于LB+SP液体培养基中,28 ℃、180 r·min-1活化12 h。将活化好的菌液以5%(体积分数)的接种量加入200 mL YSG培养基(1 L容积锥形瓶),28 ℃、150 r·min-1培养84 h。每4 h取1次样,每次吸取1 mL菌液加入比色皿中,测定不同培养时间的菌液D600值,菌株生长进入稳定期后,每12 h测1次D600值,连续测量84 h,以时间为横坐标,菌液的D600值为纵坐标。取各时间段的新鲜菌液1 mL,分别稀释10、102、103、104、105、106、107,从稀释好的菌液中吸取200 μL菌液到NBTA培养基,用玻璃棒涂布均匀后倒置于28 ℃培养箱中,待平板长出菌落,开始计数。每个稀释浓度重复3次。
1.2.2 胞外粗蛋白的制备
使用硫酸铵沉淀法沉淀粗蛋白,将硫酸铵固体研磨成粉末,加入待沉淀溶液中,至硫酸铵饱和度分别达到10%、20%、30%、40%、50%、60%、70%、80%、90%和100%。4 ℃静置6 h,12 000 r·min-1离心20 min,得到的蛋白沉淀利用缓冲液复溶后透析除盐,供后续试验使用。
1.2.3 细菌总RNA提取、质量检测和测序文库构建
分别取1 mL不同发酵时间的新鲜菌液,加入1.5 mL灭菌离心管中,4 ℃、8 000 r·min-1离心20 min去除上清液。将离心管插入液氮中速冻。每个样品重复收集3份保存于-80 ℃冰箱。样品在液氮中充分研磨破坏细菌细胞壁,加入700 μL Trizol充分混合,剧烈振荡,静置5~15 min。加入200 μL氯仿(除去蛋白)剧烈振荡,静置5 min。4 ℃、12 000 r·min-1离心15 min。小心吸取600 μL上清液(不能吸到中间层蛋白),加入等体积异丙醇(沉淀RNA)缓慢摇匀,-20 ℃静置30 min。离心机4 ℃预冷,12 000 r·min-1离心5 min,弃上清液。加入400 μL 75%(体积分数)乙醇颠倒4次清洗,12 000 r·min-1离心5 min,弃上清液,离心管倾斜放在超净台吹干,每管加入40 μL RNAase free H2O振荡摇匀,10 000 r·min-1 离心3 min。通过微量分光光度计NanoDrop测定RNA溶液的浓度和纯度。将提取的RNA迅速放置液氮中冷冻,委托北京诺禾致源科技股份有限公司完成测序和文库构建,并使用Illumina测序仪进行测序。
1.2.4 内参引物和qPCR引物设计
内参基因选择16S rRNA和ropB,利用Primer-BLAST软件,设计引物序列。引物由生工生物工程(上海)有限公司合成。内参引物序列见表1。
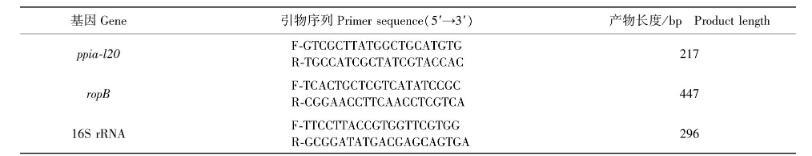
表1 RT-qPCR内参引物序列
1.2.5 测序数据分析和差异表达基因的筛选
测序数据已上传至National Center Biotechnology Information(NCBI)的SRA数据库(查询号:PRJNA1047324)。测序平台为Illumina Novaseq6000,利用Hisat 2软件将测序数据与参考基因组进行序列比对; 采用fastp软件对原始数据进行质量预处理,并对整个质控过程中的reads进行统计汇总。采用DESeq筛选差异表达基因。
1.2.6 差异表达基因的GO(gene ontology)和KEGG(Kyoto encyclopedia of genes and genomes)富集分析
富集分析采用BLAST2GO和topGOR包进行GO功能分析,得到所有显著性差异基因、上调差异基因以及下调差异基因富集的功能模块并绘图; 利用KEGG数据库对差异蛋白编码基因进行pathway分析(结合KEGG注释结果),并用超几何分布检验方法计算每个pathway条目中差异基因富集的显著性。
1.2.7 实时定量PCR分析
选取部分基因通过Real-time PCR对转录组结果进行验证,利用反转录试剂盒(ATG巨匠)对1.2.3节中获得的RNA进行反转录,得到的cDNA采用核酸定量仪Thermo Scientific NANO-Drop1000(NanoDrop Technologies,USA)进行定量,Real-time PCR靶标基因和所用引物见表2。在RNase-free离心管中配制如下混合液:RNase Free ddH2O、4 × gDNA wiper Mix、RNA template混合,用移液器轻轻吹打混匀,42 ℃加热2 min去除模板中的DNA。5 × ATGScriptTM RT Mix与反应液混合成20 μL反应体系,50 ℃反应15 min,85 ℃反应2 min。每个样本3个重复。

表2 RT-qPCR引物序列
1.3 数据统计与相关性分析
使用Prism 8.0(Graphpad)软件进行数据统计分析,所有统计数据均表示为平均值±标准差。采用Student's t-test检验, P<0.05被认为具有统计学意义。采用SPSS Statistics 26进行变量间相关性分析。两变量间相关性以Pearson相关系数r表示。
相关新闻推荐
1、青贮乳酸菌生长曲线、产酸和抑菌能力、抗生素耐药性测定(三)
2、白马耳组织成纤维细胞体外培养、冷冻前及复苏后存活率、生长曲线绘制(三)